
How to use this server
Step 1:
Click on the tab with the action you would like to perform. There are 3 options:
UPLOAD FILE- Upload your GenBank formatted file or FASTA DNA sequence file. If you upload a FASTA file containing metagenomic contigs, you may check the option directly below the "Choose File" button. The server will process your metagenomic input and return results pertaining to each contig. Please upload a multi-fasta file with this option! If a multi-fasta file is submitted without this option checked, a batch will be created with jobs for the first 10 sequences in the input file. You may view detailed INPUT INFORMATION here.
ENTER ACCESSION- Input an Accession or GI number.
PASTE SEQUENCE- Directly paste a FASTA DNA sequence in the text box. A DNA sequence may be uploaded without a header. Also, please note that IUPAC nucleotide coding is enforced for any input DNA sequence.
Click the "Run an Example" button under your section of choice if you do not have input.
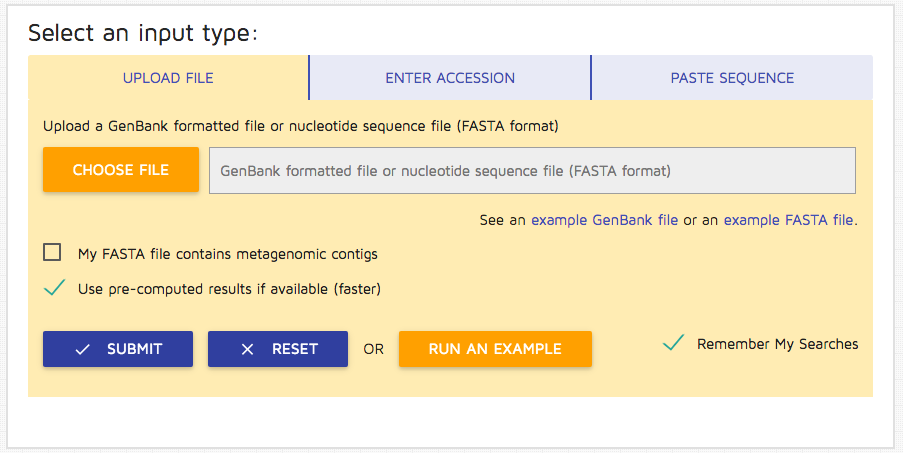
Step 2: Uncheck the option "Use pre-computed results" if you wish to run the job without retrieving existing results.
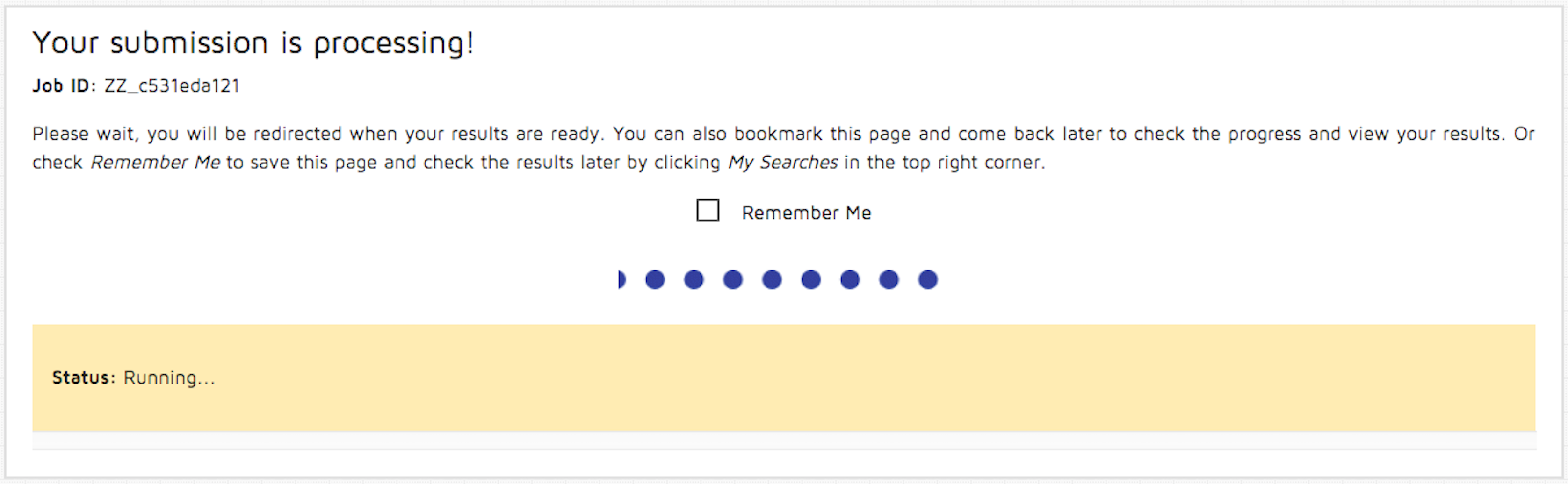
Step 3: Press the "Submit" button. The following page will be shown. You may change your 'Remember Me' status at any moment. Please note that this option requires cookies in order to retain your submission information.
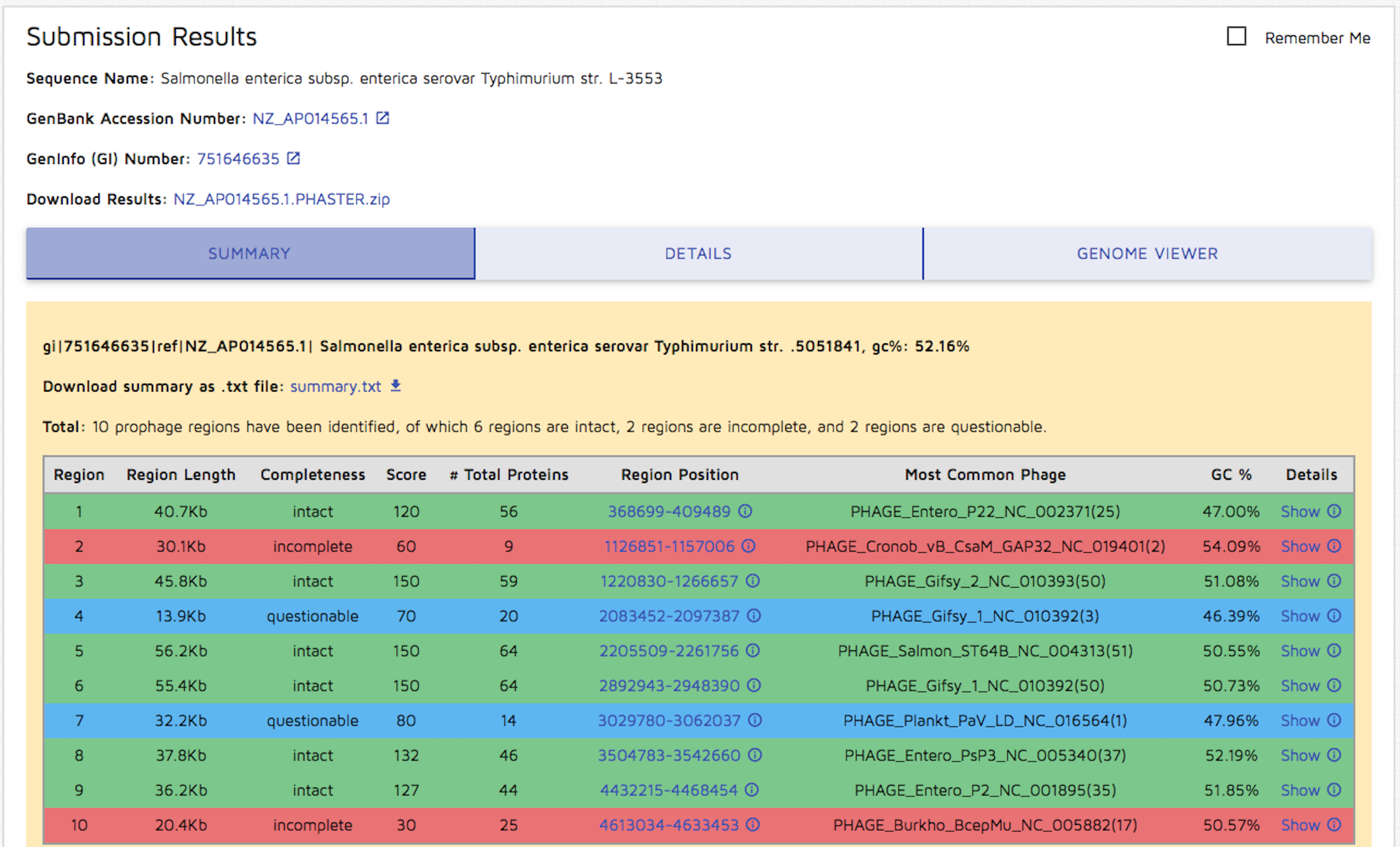
Step 4: Once the file upload has been completed, the following results page will be shown. You may download the report summary by clicking the "summary.txt" download link. The entire results package may be downloaded via the 'Download Results' link at the top of the results section. You may view detailed OUTPUT INFORMATION here.
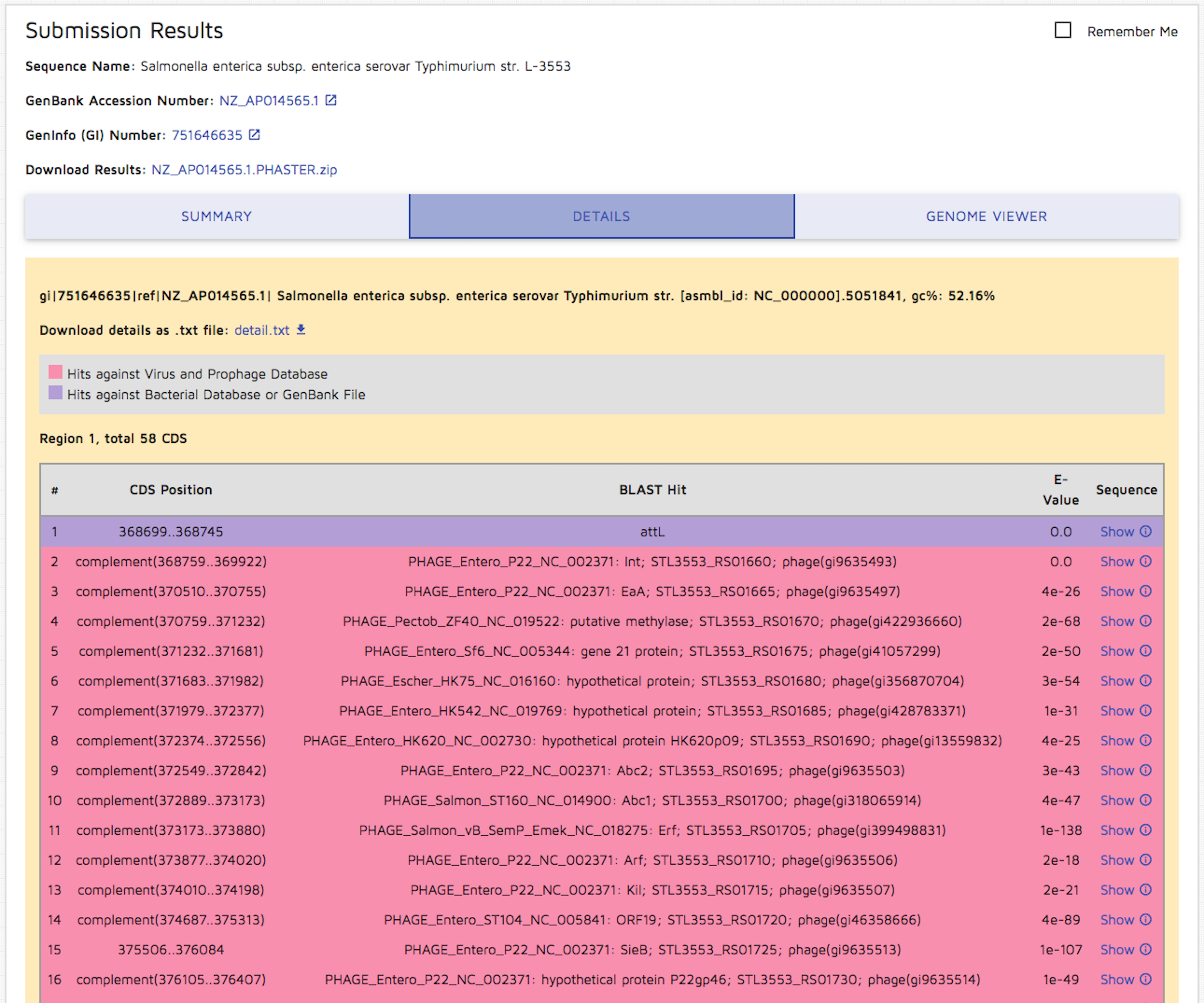
Step 5: You can view a detailed results table by clicking the "Details" tab. Again, you may download details.txt.
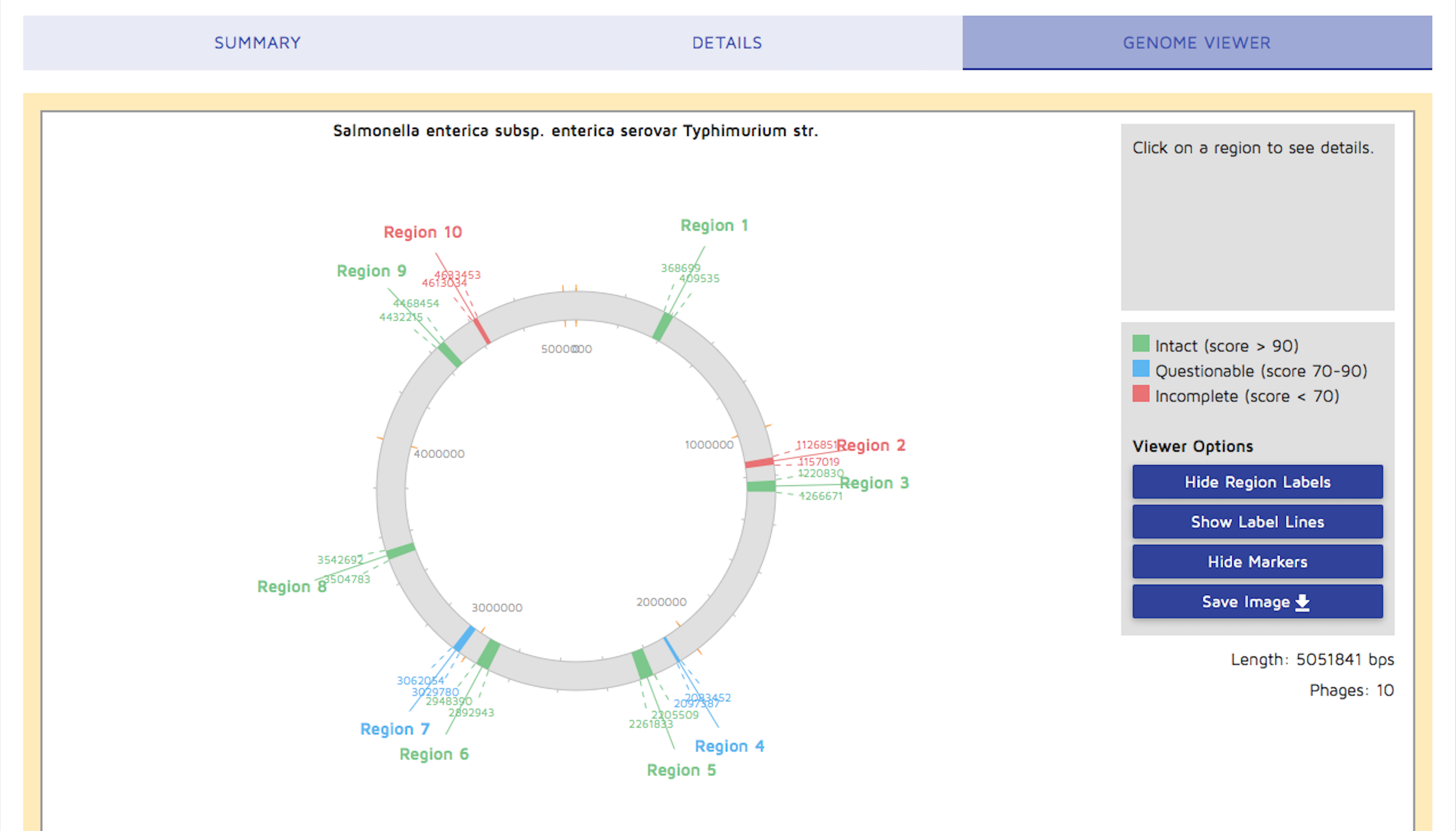
Step 6: You can view a circular genome by clicking the "Genome Viewer" tab. You may choose to display or hide the 'Region' text information on the viewer by clicking the "Hide Region Labels" button. You may also download an image of the circular genome results.
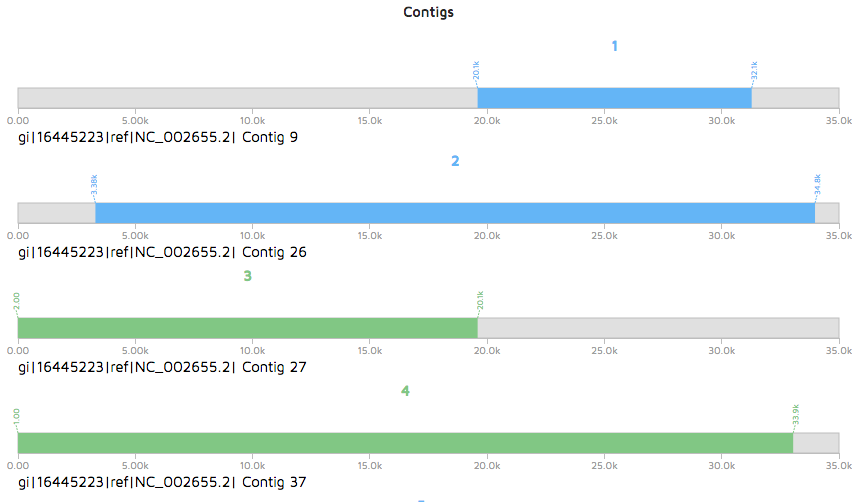
A linear view of contigs containing phage regions will be displayed instead of the circular genome if the metagenomic option was used.
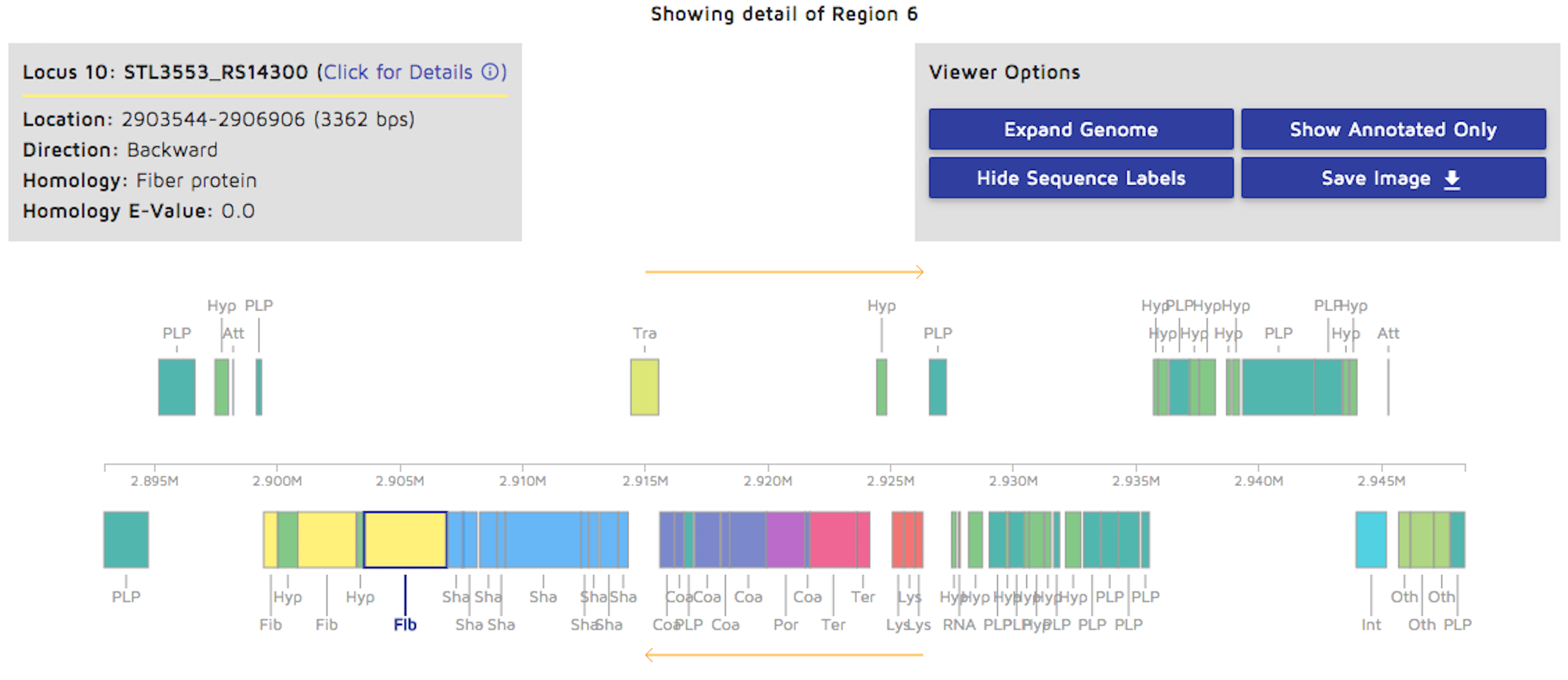
Step 7: You can view a detailed linear genome display by clicking on a region of the circular genome. You may download the resulting image as well.
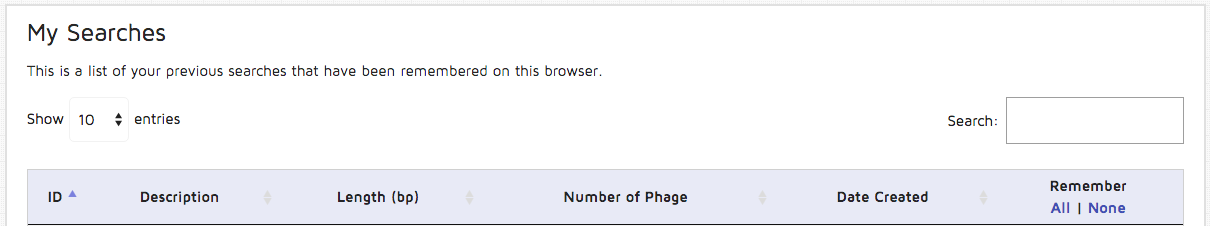
Step 8: You can view your submission history by clicking the "My Searches" option on the right side of the main header. A table with your submissions will be displayed in the following format, and you can re-view your results.
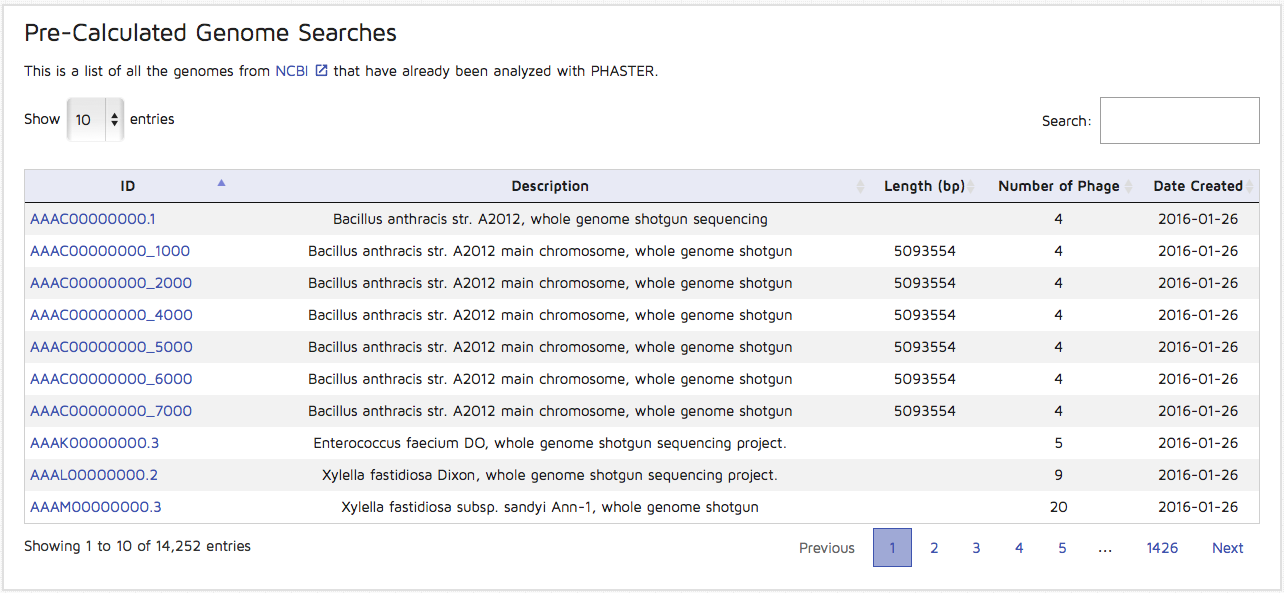
Step 9: You can view pre-calculated results for genomes by clicking the "Genomes" option on the right side of the main header. You may browse through the paginated list or search for a particular GI or Accession number in order to view results wihtout running a new submission.
How to use the URLAPI
PHASTER's URLAPI may be easily accessed or integrated into a user's local program. You can use any html request tool for Windows, Linux, or Mac, such as wget, to connect to our server using the following URLAPI's. The characters '<;>' denote user input.
1. POST to phaster.ca/phaster_api - Upload a FASTA DNA sequence (minimum length is 1500 bp). Please note that the maximum file size is 26Mb. Please submit one sequence at a time. Other input formating/restrictions apply - INPUT INFORMATIONResponse:
A JSON object with the following fields:
- 'job_id' - The Job ID of your new submission.
- 'status' - The status of your new submission. This may include the position of the job in the queue if the job has not started to run yet.
- 'error' - This field is included if there are initial input problems and the job is not queued.
Example: 'wget --post-file="example.fna" "http://phaster.ca/phaster_api" -O Output_filename'
Response: {"job_id":"ZZ_023a167bf8","status":"You're next!..."}
2.POST to phaster.ca/phaster_api with a multicontig file in Multi-FASTA format - Upload at least two FASTA DNA contigs with minimum length of 2000 bp each. Make sure to set the 'contigs' option to 1. Other input formating/restrictions apply - INPUT INFORMATION
Example: 'wget --post-file="contigs.fna" "http://phaster.ca/phaster_api?contigs=1" -O Output_filename'
Response: {"job_id":"ZZ_023a167bf8","status":"You're next!..."}
3. GET phaster.ca/phaster_api?acc=<;> - Input Accession number, GI number, or Job ID.
PHASTER is run for the given Accession or GI number (or an existing result retrieved). The status and/or the results are returned. If a Job ID for a submission is given, the status for that submission is returned. The results are returned if the job is complete.
Response:
A JSON object with the following fields:
- 'job_id' - The Accession number, GI number, or Job ID of the submission.
- 'status' - The status of the submission. This may include the position of the job in the queue if the job has not started to run yet.
- 'url' - This field is included if the submission completed successfully. The given url may be used to view the results using the web interface.
- 'zip' - This field is included if the submission completed successfully. The given url may be used to download the available result files.
- 'summary' - This field is included if the submission completed successfully. The summary of the results is retrieved.
- 'error' - This field is included if there are initial input problems, or if the submission failed.
Example: 'wget "http://phaster.ca/phaster_api?acc=NC_000913" -O Output_filename'
Response: {"job_id":"NC_000913.3","status":"Complete","url":"phaster.ca/submissions/NC_000913.3",
"zip":"phaster.ca/submissions/NC_000913.3.zip","summary":"Criteria for scoring prophage regions..."}"
Example: 'wget "http://phaster.ca/phaster_api?acc=ZZ_023a167bf8" -O Output_filename'
Response: {"job_id":"ZZ_023a167bf8","status":"Running..."}